Welcome to the documentation page for SCOT!
SCOT is an unsupervised algorithm for performing cell-to-cell alignment of single-cell multi-omic datasets.
We provide tutorials and examples on this website. For details on methodology and experimental results, please check out our pre-print on bioRxiv
Overview
Availability of various sequencing technologies allows us to capture different properties of the genome at the single-cell resolution and get a holistic view of the single-cell genome. However, with very few exceptions of co-assaying technologies,applying different sequencing assays on the same single-cell is not possible.
In such situations, we can rely on computational tools to align multi-omic data from separate but related datasets (e.g. aliquots of the same population). As long as the measurements are expected to have some shared underlying biological manifold (e.g. taken from cell populations that contain the same cell types/states or share geneology), we expect to have sample-wise correspondence probabilities to recover across measurements and be able to align these datasets for an integrated view.
This problem requires unsupervised computational methods because the process yields disparate datasets: Since we measure different properties of the genome on different cells, we have no 1-to-1 correspondence information between either the cells or the features.

SCOT is an unsupervised alignment tool that yields correspondence probabilities between cells from different -omics datasets. Unlike many other integration methods, it runs without requiring any correspondence information a priori and aligns datasets based on the correspondence probabilities it recovers.
SCOT works in three steps:
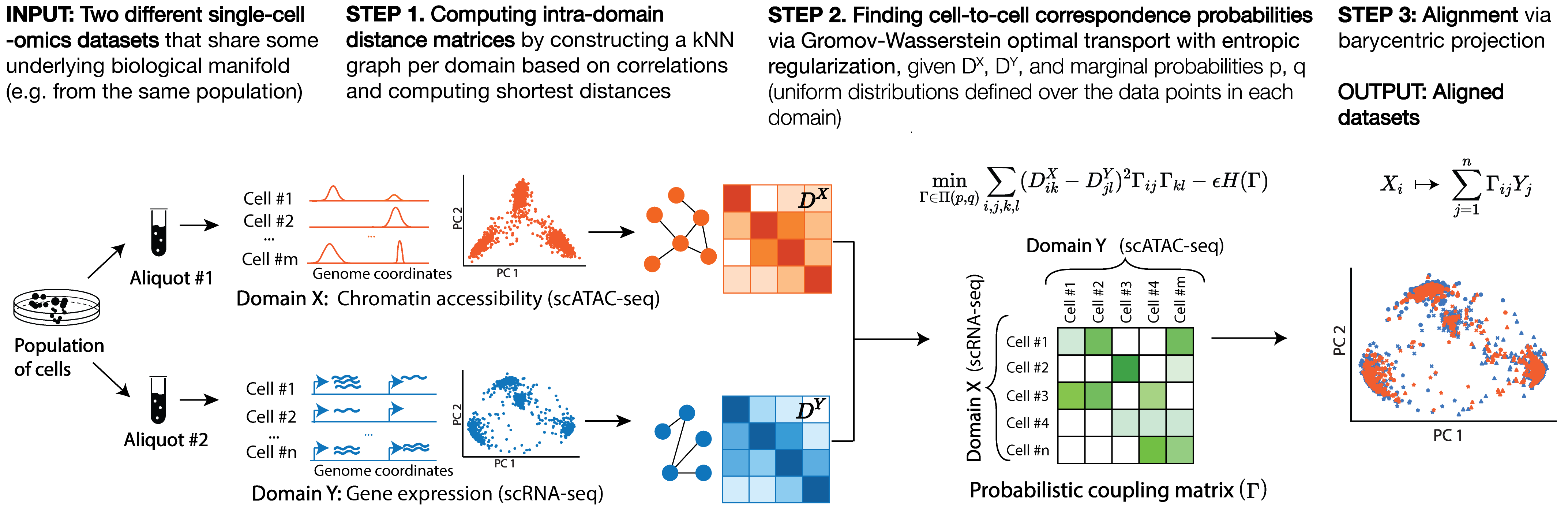
Why should I prefer to use SCOT on my datasets?
While there are other alignment tools available, SCOT brings a few advantages that are important in real-world settings:
1. High quality multi-omic alignment
Most alignment methods are developed for batch integration of single-cell RNA-seq datasets (e.g. scAlign, MNN, Seurat, Harmony, LIGER) and are shown to perform poorly on multi-omic alignment tasks, which is fundamentally a different problem. SCOT is specifically designed for and tested on multi-omic integration tasks.
2. Unsupervised alignment
Many alignment tools (e.g. MAGAN, Seurat) require anchor points or feature-wise correspondence information to perform alignment, which one does not likely to have upon sequencing separate cell populations. SCOT is an unsupervised tool and does not require any correspondence information to be known a priori in order to perform alignment.
3. Approximately self-tuning hyperparameters
Other currently available unsupervised multi-omic alignment tools (UnionCom, MMD-MA, Pamona) require users to perform hyperparameter optimization in order to yield high quality alignments. Without any validation data on correspondences, it is difficult to perform hyperparameter tuning. SCOT provides a procedure to approximately self-tune hyperparameters in fully unsupervised settings.
4. Handling cell type imbalance
Through an extension with unbalanced optimal transport, SCOT is able to handle cell type imbalance between multi-omic assays.
5. Computational scalability
In comparison to the other unsupervised mutli-omic alignment tools, SCOT is computationally scalable to large datasets.
Contact and Citation
If you would like to use SCOT for your datasets, take a look at the tutorial page, as well as the examples we provide on this documentation site. If you are having trouble getting started or have questions about SCOT, please do not hesitate to contact us!
If you use SCOT for your work, you can cite our pre-print as below:
BibTex Citation:
@article {SCOT2020,
author = {Demetci, Pinar and Santorella, Rebecca and Sandstede, Bj{\"o}rn and Noble, William Stafford and Singh, Ritambhara},
title = {Gromov-Wasserstein optimal transport to align single-cell multi-omics data},
elocation-id = {2020.04.28.066787},
year = {2020},
doi = {10.1101/2020.04.28.066787},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2020/11/11/2020.04.28.066787},
eprint = {https://www.biorxiv.org/content/early/2020/11/11/2020.04.28.066787.full.pdf},
journal = {bioRxiv}.
}